Rapidly mutating DNA regions were mapped using a multi-generational family and advanced sequencing tools.
Understanding how human DNA changes over generations is crucial for estimating genetic disease risks and tracing our evolutionary history. However, some of the most variable regions of our DNA have been difficult for researchers to access, until now.
Scientists from University of Utah Health, the University of Washington, PacBio, and other institutions have used advanced DNA sequencing technologies to create the most detailed map of genetic change across generations. Their study revealed that certain parts of the human genome change far more rapidly than previously believed, opening the door to deeper insights into the origins of human disease and evolution.
"It's mutations that ultimately differentiate us from other species," says author Lynn Jorde, PhD. "We're getting at a very basic property of what makes us human."
The results are published in the journal Nature.
The biological "speed of light"
By comparing the genomes of parents and their children, the team was able to measure how frequently new mutations occur and are passed down. Jorde explains that this mutation rate is as fundamental to human biology as the speed of light is to physics. "This is something you really need to know—the speed at which variation comes into our species," says Jorde, professor of human genetics at the Spencer Fox Eccles School of Medicine at the University of Utah. "All of the genetic variation that we see from individual to individual is a result of these mutations." Over generations, these changes have produced everything from differences in eye color to the ability to digest lactose to rare genetic disorders.

The researchers estimate that every human has nearly 200 new genetic changes that are different from either parent. Many of these changes occur in regions of DNA that are especially difficult to study.
Aaron Quinlan, PhD, professor and chair of human genetics in SFESOM and an author on the study, says that previous efforts to study human genetic change were limited to the parts of the genome that mutate the least. But the new study used advanced sequencing technologies to reveal the most rapidly changing regions of human DNA—regions that Quinlan describes as "previously untouchable."
"We saw parts of our genome that are crazy mutable, almost a mutation every generation," he says. Other segments of DNA were more stable.
Jorde says that the new resource can be an important support for genetic counseling by helping answer the question, "If you have a child who's affected with a disease, is it likely to be inherited from a parent, or is it likely to be a new mutation?" Diseases caused by changes in "mutation hotspots" are more likely to be unique to the child, rather than having been passed down from their parents. This means that the risk of the parents having other kids with the same disease is lower. But if a genetic change was inherited from the parents, those parents' future kids have a higher risk of having the disease.
The platinum pedigree
The researchers' discovery hinged on a Utah family that has worked with genetics researchers since the 1980s as part of the Centre d'Etude du Polymorphisme Humain consortium, proving invaluable for the Human Genome Project.
Four generations of the family have donated DNA and consented to its analysis, which allowed the researchers an extraordinarily in-depth look at how new changes arise and are inherited from parents to children. "A large family with this breadth and depth is an incredibly unique and valuable resource," says Deborah Neklason, PhD, research associate professor of internal medicine in SFESOM and an author on the study. "It helps us understand variation and changes to the genome over generations in incredible detail."
To get a complete, high-resolution picture of genetic variation over time, the team sequenced each person's DNA using multiple different technologies. Some technologies are best for detecting the smallest possible changes to DNA; others can scan enormous swaths of DNA at a time to find big changes and see parts of the genome that are otherwise difficult to sequence. By sequencing the same genomes with multiple technologies, the researchers achieved the best of both worlds: accuracy on both a small and large scale.
In future work, the researchers hope to extend their comprehensive sequencing techniques to more people to see if the genetic rate of change is different for different families. "We saw really interesting stuff in this one family," Quinlan says. The next question is, "How generalizable are those findings across families when trying to predict risk for disease or how genomes evolve?"
The sequencing results will be made freely available so that other researchers can use the data in their own studies, opening the door to further insights into human evolution and genetic disease.
Reference: "Human de novo mutation rates from a four-generation pedigree reference" by David Porubsky, Harriet Dashnow, Thomas A. Sasani, Glennis A. Logsdon, Pille Hallast, Michelle D. Noyes, Zev N. Kronenberg, Tom Mokveld, Nidhi Koundinya, Cillian Nolan, Cody J. Steely, Andrea Guarracino, Egor Dolzhenko, William T. Harvey, William J. Rowell, Kirill Grigorev, Thomas J. Nicholas, Michael E. Goldberg, Keisuke K. Oshima, Jiadong Lin, Peter Ebert, W. Scott Watkins, Tiffany Y. Leung, Vincent C. T. Hanlon, Sean McGee, Brent S. Pedersen, Hannah C. Happ, Hyeonsoo Jeong, Katherine M. Munson, Kendra Hoekzema, Daniel D. Chan, Yanni Wang, Jordan Knuth, Gage H. Garcia, Cairbre Fanslow, Christine Lambert, Charles Lee, Joshua D. Smith, Shawn Levy, Christopher E. Mason, Erik Garrison, Peter M. Lansdorp, Deborah W. Neklason, Lynn B. Jorde, Aaron R. Quinlan, Michael A. Eberle and Evan E. Eichler, 23 April 2025, Nature.
The work was supported by funding from the National Institutes of Health (grant numbers R01HG002385, R01HG010169, U24HG007497, 5K99HG012796-02, R00HG011657, R35GM118335, and GM147352), the Terry Fox Research Foundation (grant number 1074), and the Canadian Institutes of Health Research (grant number 159787).
Researchers report the following conflicts of interest: Evan Eichler is a scientific advisory board (SAB) member of Variant Bio, Inc. Charles Lee is an SAB member of Nabsys and Genome Insight. David Porubsky has previously disclosed a patent application (no. EP19169090) relevant to Strand-seq. Zev Kronenberg, Cillian Nolan, Egor Dolzhenko, Cairbre Fanslow, Christine Lambert, Tom Mokveld, William Rowell, and Michael Eberle are employees and shareholders of PacBio. Zev Kronenberg is a private shareholder in Phase Genomics. The other authors declare no competing interests.
News

Why music from your youth still has such an intense effect years later: A psychological perspective
You're driving, and suddenly a familiar song fills the air. Before you even know it, a wave of emotions comes over you – not just memories, but a deep, almost physical feeling. This powerful [...]
AI to antibody in days: breaking the wet lab bottleneck via high-throughput integration
The role of artificial intelligence (AI) in drug design has fundamentally shifted from a speculative tool to a central pillar of pharmaceutical research and development (R&D). Sino Biological plays a critical role in this [...]

Regenerative Healthcare by Design: Engineering Health-Centric Buildings and Urban Ecosystems
Introduction The next evolution of healthcare will not be confined to hospitals, clinics, or episodic interventions—it will be embedded into the infrastructure of everyday life. Regenerative health ecosystems require a systemic re-architecture of how [...]

Scientists Warn: Humanity Has Pushed the Planet Past Its Limits
Human population and consumption have surpassed Earth’s limits, increasing risks to climate and global stability. The Earth is already operating beyond its capacity to sustainably support the global population, according to new research highlighting [...]
Breakthrough Study Reveals Why Damaged Nerves Struggle To Heal
A newly identified molecular mechanism reveals how neurons weigh survival against repair after injury. Scientists at the Icahn School of Medicine at Mount Sinai have identified a molecular switch in neurons that limits the regrowth of [...]
Popular Vitamin B3 Supplements May Help Cancer Cells Survive, Scientists Warn
A new study raises important questions about widely used NAD+ supplements, suggesting that compounds often taken to boost energy and support healthy aging may have unintended consequences in cancer treatment. Millions of Americans take [...]
Scientists Discover Cancer Tumors Are “Addicted” to This Common Antioxidant
Cancer cells may be exploiting a common antioxidant as fuel, revealing a potential weakness that future therapies could target. Cancer cells may be tapping into an unexpected energy source: an antioxidant long associated with [...]
Nanotube injector transfers cytoplasmic contents and organelles between living cells safely
Cells are not isolated units; they continuously exchange proteins, genetic material, and even entire organelles with their neighbors. Intercellular transfer influences how tissues develop, respond to stress, and repair damage. In certain cancers, for [...]

CEO of America’s largest public hospital system is ready to replace radiologists with AI
The chief executive of America’s largest public hospital system says he is prepared to start replacing radiologists with artificial intelligence in some circumstances, once the regulatory landscape catches up. Mitchell H. Katz, MD, president [...]

Our books now available worldwide!
Online Sellers other than Amazon, Routledge, and IOPP Indigo Global Health Care Equivalency in the Age of Nanotechnology, Nanomedicine and Artifcial Intelligence Global Health Care Equivalency In The Age Of Nanotechnology, Nanomedicine And Artificial [...]

Study finds higher heart disease risk in long COVID patients
People with long COVID are at increased risk of developing cardiovascular disease, according to a new study from Karolinska Institutet published in eClinicalMedicine. The results show that the risk of conditions such as cardiac arrhythmias [...]
The Corona variant Cicada is here – we know that
Online and on social media, reports are piling up about a new Sars-Cov-2 variant that is currently on the rise: BA.3.2, also known as Cicada. That's what it's all about: The Omicron variant BA.3.2, [...]

A Simple Blood Test Could Predict Dementia Risk 25 Years Early
A single blood marker may quietly signal dementia risk decades in advance. Scientists at the University of California, San Diego, have identified a blood signal that could forecast dementia risk decades before symptoms begin. Their [...]
Sperm Get Lost in Space and Scientists Finally Know Why
Having a baby in space may be far more complicated than expected, as new research shows sperm struggle to find their way in microgravity. Starting a family beyond Earth could be more complicated than [...]
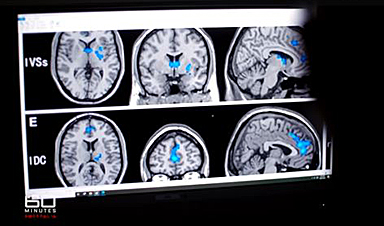
Digital Dementia – Brain fog and disassociation from being chronically online
New medical evidence, featured on 60 Minutes Australia, indicates excessive screen time is causing "digital dementia" in young Australians, with brain scans showing physical shrinkage and damage. Experts warn that high device usage (6-8 hours [...]
A new, highly mutated COVID variant called ‘Cicada’ is spreading in the US.
BA.3.2, a heavily mutated new COVID-19 variant which may be better able to escape immunity from vaccines or prior infection, is now spreading in the United States. Although COVID cases are currently low nationally, [...]