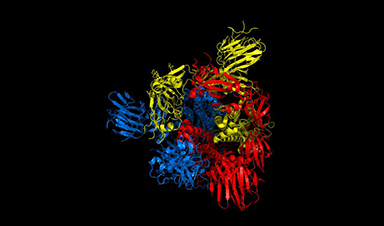
esearchers have detailed a mechanism in the distinctive corona of Covid-19 that could help scientists to rapidly find new treatments for the virus, and quickly test whether existing treatments are likely to work with mutated versions as they develop.
The team, led by the University of Warwick as part of the EUTOPIA community of European universities, have simulated movements in nearly 300 protein structures of the Covid-19 virus spike protein by using computational modelling techniques, in an effort to help identify promising drug targets for the virus.
In a new paper published today (19 February) in the journal Scientific Reports, the team of physicists and life scientists detail the methods they used to model the flexibility and dynamics of all 287 protein structures for the Covid-19 virus, also known as SARS-CoV-2, identified so far. Just like organisms, viruses are composed of proteins, large biomolecules that perform a variety of functions. The scientists believe that one method for treating the virus could be interfering with the mobility of those proteins.
They have made their data, movies and structural information, detailing how the proteins move and how they deform, for all 287 protein structures for Covid-19 that were available at the time of the study, publicly accessible to allow others to investigate potential avenues for treatments.
The researchers focused particular efforts on a part of the virus known as the spike protein, also called the Covid-19 echo domain structure, which forms the extended corona that gives coronaviruses their name. This spike is what allows the virus to attach itself to the ACE2 enzyme in human cell membranes, through which it causes the symptoms of Covid-19.
The spike protein is in fact a homotrimer, or three of the same type of protein combined. By modelling the movements of the proteins in the spike, the researchers identified a ‘hinge’ mechanism that allows the spike to hook onto a cell, and also opens up a tunnel in the virus that is a likely means of delivering the infection to the hooked cell. The scientists suggest that by finding a suitable molecule to block the mechanism – literally, by inserting a suitably sized and shaped molecule – pharmaceutical scientists will be able to quickly identify existing drugs that could be effective against the virus.
Lead author Professor Rudolf Roemer from the Department of Physics at the University of Warwick, who conducted the work while on a sabbatical at CY Cergy-Paris Université, said: “Knowing how this mechanism works is one way in which you can stop the virus, and in our study we are the first to see the detailed movement of opening. Now that you know what the range of this movement is, you can figure out what can block it.
“All those people who are interested in checking whether the protein structures in the virus could be drug targets should be able to examine this and see if the dynamics that we compute are useful to them.
“We couldn’t look closely at all the 287 proteins though in the time available. People should use the motion that we observe as a starting point for their own development of drug targets. If you find an interesting motion for a particular protein structure in our data, you can use that as the basis for further modelling or experimental studies.”
To investigate the proteins’ movements, the scientists used a protein flexibility modelling approach…
Image Credit: University of Warwick
Post by Amanda Scott, NA CEO. Follow her on twitter @tantriclens
Thanks to Heinz V. Hoenen. Follow him on twitter: @HeinzVHoenen
News
Why More People in Their 30s Are Suddenly Getting Colon Cancer
A major Swiss study found that colorectal cancer is becoming increasingly common in adults under 50, even as rates decline in older age groups. Researchers in Switzerland have identified a concerning trend: while colorectal [...]
Researchers Compare MS Models to Human Tissue in Search for Better Therapies
Researchers identified key differences between two widely used multiple sclerosis models, showing how each can better study myelin damage, immune responses, and repair. The findings may improve efforts to develop treatments that restore lost [...]
Scientists Discover Genetic “Off Switch” That Supercharges CAR T Cells Against Cancer
A new study reveals a possible way to make CAR T-cell therapy more durable and effective by targeting a single gene-regulating protein. CAR T-cell therapy is widely seen as a breakthrough in personalized cancer [...]
New Vitamin B12-Based Therapy Could Change How Brain Cancer Is Treated
Researchers have identified a vitamin B12–based compound that appears capable of crossing the blood–brain barrier and selectively accumulating in glioblastoma tissue. For decades, one of the biggest problems in brain cancer treatment has had [...]

Simple Fiber Supplement Cuts Knee Arthritis Pain in Just 6 Weeks, Study Finds
A daily inulin supplement may help reduce knee osteoarthritis pain while revealing a possible link between gut health, muscle function, and pain sensitivity. For millions of people living with knee osteoarthritis, managing chronic pain [...]
This Common Vitamin May Help Stop Prediabetes From Turning Into Diabetes
Vitamin D may help prevent type 2 diabetes in people with specific genetic variations, offering a possible path toward personalized diabetes prevention. More than 40% of U.S. adults have prediabetes, a condition in which [...]

Ebola, hantavirus: Is the world prepared for the next pandemic?
Funding cuts to health research and a growing antivaccine movement are making it harder than ever to respond to viruses. The World Health Organization (WHO) has declared that an Ebola outbreak in Uganda and [...]

May 2026 Healthcare News and Trends: Market Signals That Matter
Artificial intelligence is dominating headlines, telehealth has settled into a new normal, and digital health continues to promise transformation. However, much of what is being discussed in healthcare today reflects potential rather than reality. [...]
Scientists Rewire Donor Stem Cells To Outsmart Aggressive Blood Cancers
Researchers have tested a gene-edited stem cell transplant designed to shield healthy blood-forming cells from powerful cancer-targeting immunotherapies. For patients with highly aggressive blood cancers, stem cell transplantation can offer a rare chance at [...]

Recent Digital Health Trends, Insights and News – May 2026
Last month marked continued progress as digital health moves into its next phase — from AI expanding into drug discovery and core infrastructure to new federal pathways accelerating device access and home-based care. Together, [...]
Cancer Mystery Solved: Scientists Discover How Melanoma Becomes “Immortal”
Scientists have uncovered a previously overlooked mechanism that may help melanoma cells become effectively “immortal.” Cancer cells face a major problem before they can become deadly: They have to figure out how to stop [...]
How Visual Neurons Organize Thousands of Synaptic Inputs
Summary: A new study uncovered the organizational rules that determine how neurons in the primary visual cortex process information. By imaging both the cell bodies (soma) and the individual synapses (on dendritic spines) of [...]

Scientists Just Found a Surprising Way To Destroy “Forever Chemicals”
Scientists have uncovered a new mechanism that may help break down highly persistent PFAS pollutants. PFAS have earned the nickname “forever chemicals” for a reason. These industrial compounds are so chemically durable that they [...]
Scientists Discover Cheap Material That Kills Deadly Superbugs
A new sulfur-rich antimicrobial polymer shows strong effectiveness against fungal and bacterial pathogens and may offer an affordable solution to antimicrobial resistance. Antimicrobial resistance is creating growing challenges for both healthcare and food production, [...]

What to Know About Cicada, or BA.3.2, the Latest SARS-CoV-2 Variant Under Monitoring
Like periodical cicadas, the insects for which it is nicknamed, SARS-CoV-2 Omicron subvariant BA.3.2 is only just beginning to emerge after lying low for an extended period since it first appeared. Although it was [...]
Scientists Say This Simple Supplement May Actually Reverse Heart Disease
Scientists in Japan say a common supplement may actually help “unclog” certain diseased heart arteries from the inside out. A simple food supplement sold in Japan may have helped reverse a dangerous form of [...]