Cancer is a complex and diverse disease, and its range of associated mutations is vast. The combination of these genomic changes in an individual is referred to as their "mutational landscape." These landscapes vary based on the type of cancer, and even individuals with the same type of cancer can have very different mutation patterns.
Scientists have already documented the mutational landscapes of several forms of cancer. Somatic structural variations (SVs) have been found to account for over 50% of all cancer-causing mutations. These mutations occur in cells over time, such as through copying errors in DNA during cell division, resulting in alterations to the chromosome structure.
They are not inherited and are found only in affected cells and in their daughter cells. As we age, such genomic alterations become more numerous, and a person's mutational landscape increasingly comes to resemble a unique mosaic.
"We developed a computational analysis method to detect and identify the functional effects of somatic SVs," she reports. This enabled the team to understand the molecular consequences of individual somatic mutations in different leukemia patients, giving them new insights into the mutation-specific alterations. Sanders says it may also be possible to use these findings to develop therapies that target the mutated cells, adding that "they open up exciting new avenues for personalized medicine."
Even more detailed than conventional single-cell analyses
Their calculations are based on data from Strand-seq – a special single-cell sequencing method that Sanders played an instrumental role in developing and that was first introduced to the scientific community in 2012. This technique can examine a cell's genome in much greater detail than conventional single-cell sequencing technologies. Thanks to a sophisticated experimental protocol, the Strand-seq method can independently analyze the two parental DNA strands (one from the father and one from the mother).
With conventional sequencing methods, distinguishing such homologs – chromosomes that are similar in shape and structure but not identical – is nearly impossible. "By resolving the individual homologs within a cell, somatic SVs can be identified much better than with other methods," explains Sanders. The approach used for doing this was described by the researcher and her colleagues in a paper that appeared in Nature Biotechnology in 2020.
The research team is part of the joint research focus "Single-Cell Approaches for Personalized Medicine" of the Berlin Institute of Health at Charité (BIH), Charité – Universitätsmedizin Berlin, and the Max Delbrück Center.
Building on this work, they are now able to also determine the positions of nucleosomes in each cell. Nucleosomes are units of DNA wrapped around protein complexes called histones, and play a crucial role in organizing chromosomes. The position of nucleosomes can change during gene expression, with the type of wrapping revealing whether or not a gene is active. Sanders and her colleagues developed a self-learning algorithm to compare the gene activity of patient cells with and without somatic SV mutations, allowing them to determine the molecular impact of the structural variants.
New targets for cancer therapy
"We can now take a sample from a patient, look for the mutations that led to the disease, and also learn the signaling pathways that the disease-causing mutations disrupt," explains Sanders. For example, the team was able to identify a rare but very aggressive mutation in a leukemia patient. The nucleosome analysis provided the researchers with information about the signaling pathways involved, which they used to specifically inhibit the growth of cells containing the mutation. "This means that a single test tells us something about the cellular mechanisms involved in cancer formation," says Sanders. "We can eventually use this knowledge to develop personalized treatments, guided by each patient's unique condition."
News

Paralyzed Man Feels Sensation Again With Brain Stimulation Device
Aneuroprosthetic system has allowed a man with paralysis to grasp and lift objects and feel touch again. The device helped 42-year-old Keith Thomas of Massapequa, New York, who was paralyzed from the chest down [...]
Global Cancer Cases Could Surge 67% by 2050, New Report Warns
New data reveal major geographic disparities and highlight the urgent need for global action on prevention, early detection, and equitable access to treatment. For roughly one in five people worldwide, cancer will become part [...]
A Deadly Ebola-Like Virus Is Spreading. Are We Ready?
BU virologist Nancy Sullivan says the Bundibugyo outbreak in the Democratic Republic of the Congo underscores the need for broader outbreak preparedness. The death of a nurse marked the moment health officials recognized that [...]

Why Most Animal Viruses Never Become Human Pandemics
From receptor mismatch to risky human-animal interfaces, this article explains why spillover is common but true pandemic emergence remains rare. Introduction Humans are constantly exposed to animal viruses through farming, wildlife contact, and the [...]

Stem cell organoids repair heart microvessels in coronary artery disease models
A Stanford University team has shown that vascular organoids derived from human stem cells can repair the heart’s microvessel network in pigs with ischaemic heart disease – a proof-of-concept advancement that could open new therapeutic [...]

Goodbye GP waiting rooms, hello prevention at home
Prevention is suddenly everywhere in NHS reform. The recent £340m community pharmacy deal is moving more services onto the high street. Community Diagnostic Centres are being expanded, and the Neighbourhood Health Framework sets out [...]
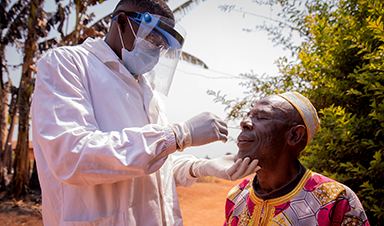
Ebola control is weakened by mistrust and cultural insensitivity
Effective response depends on cooperation with communities and frontline workers, writes Zaeem ul Haq The current Bundibugyo Ebola outbreak in the Democratic Republic of the Congo (DRC) and Uganda is exposing dangerous gaps in [...]

Building the Brain Requires Millions of Dangerous DNA Breaks
Scientists discovered that building a healthy brain involves an unexpected step: young neurons routinely break and rapidly repair their own DNA. As the brain develops, newly formed nerve cells must travel through tightly packed tissue [...]

One Tiny Change May Explain How Viruses Jump From Bats to Humans
Scientists found that one tiny genetic change may determine whether a bat virus stays in bats or becomes a human threat. Most infectious disease outbreaks begin when a virus or other pathogen crosses from animals into [...]

Scientists Discover 250+ Genes That Could Lead to New Ways To Prevent Melanoma
The world’s largest study of mole genetics identified hundreds of genes tied to melanoma risk, uncovering potential new drug targets and paving the way for more accurate melanoma screening and prevention. Researchers at QIMR [...]
Breakthrough Diabetes Treatment Reprograms the Immune System
An engineered stem cell therapy reversed new-onset Type 1 diabetes in mice by shifting the immune system away from attacking insulin-producing cells. For more than a century, people with Type 1 diabetes have relied [...]
Taking the world’s temperature: WHO chief spotlights global health emergencies
Taking the world’s temperature on pressing health matters, WHO Director-General Tedros Adhanom Ghebreyesus provided the latest on current global challenges - and successes when it comes to international cooperation. “The outbreaks of hantavirus, Ebola and Marburg all show [...]
Scientists Create Tiny “Mini Livers” That Could One Day Replace Liver Transplants
Engineered tissue grafts could help perform key liver functions and benefit thousands of people living with liver failure. The liver is one of the body’s hardest-working organs, carrying out hundreds of vital jobs, from [...]

NanoMedical Brain/Cloud Interface – Explorations and Implications. A new book from Frank Boehm
New book from Frank Boehm, NanoappsMedical Inc Founder: This book explores the future hypothetical possibility that the cerebral cortex of the human brain might be seamlessly, safely, and securely connected with the Cloud via [...]
Scientists Discover Surprising Way To Help the Brain Recover After Stroke
A new study suggests that strengthening the body’s natural circadian rhythms may help the brain recover after stroke, even when treatment begins days after the injury. Every year, millions of people survive a stroke, [...]

Our books now available worldwide!
Online Sellers other than Amazon, Routledge, and IOPP Indigo Global Health Care Equivalency in the Age of Nanotechnology, Nanomedicine and Artifcial Intelligence Global Health Care Equivalency In The Age Of Nanotechnology, Nanomedicine And Artificial [...]